Quick user guide
The pipeline is configured by default to perform a functional differential expression analysis with paired-end FASTQ files without barcodes. All modules and some steps of the pipeline can be disabled individually adding these options:
-nqc (no quality control)
-nrrnarem (no rRNA removal module)
-nfunmap (no mapping of reads to functional databases)
-nsum (don’t create abundance table)
-nde (don’t perform a differential expression analysis with DESeq2)
However, be aware when disabling modules, bear in mind that the output of each module is passed to the following.Therefore you must be sure that if you disable a module, the input for the next module is previously processed.
For the functional analysis, the MetaHIT-2014 gene catalog database is used by default. To use the “M5nr” database this options must be supplied “-m5nr”.
To perform a taxonomical analysis, two options might be used :
-tax (map read to taxonomical databases)
-sumtax (create taxonomical abundance table)
A full description of the pipeline can be found here.
If input is not paired-end data, additional customization of the pipeline will be required. Its implementation has been done following POSIX compatibility and therefore should run in any unix-like shell (list of differences between the common login shell “bash” and the Ubuntu POSIX system shell “dash” can be found here). Furthermore, the pipeline design is simple; it is based on a chaining of inputs and outputs between modules and tasks within each module.
Installation
The pipeline is split into three files (corresponding to pipeline scripts, third-party tools and databases) that must be downloaded and unpacked in the same folder. Please, ensure the folder name does not contain whitespaces.
tar xvf 1-Scripts.tar.gz
tar xvf 2-ThirdpartyTools.tar.gz
tar xvf 3-Databases.tar.gz
To configure proper paths to system shell in all scripts, just run this command in the pipeline’s root folder where you unpacked all files (there is no need to compile):
./config
Input
All sequenced paired-end files in FASTQ format from all the samples must be placed here:
./0-Sequences
Two files must be properly configured before running the pipeline. Two links in the pipeline’s root folder are automatically created after running “./config” to access them:
Content example:
 Metadata file example
Metadata file example
These columns must be specified to properly give an analysis context (white spaces are not allowed). Other columns can be added to describe the study, but won't be used by the pipeline:
SampleName
Specify the sample names that will be used to generate intermediate results.
SampleFiles
Two filenames, separated by comma, specifying the two pair-end FASTQ files to be used for each sample. All FASTQ files must be placed into “./0-Sequences” folder.
Shortname
Sample shortnames to be used in results files.
Order
Integer number to specify the order in which samples will be processed. To disable samples to be processed by the pipeline use number 0.
Fx-<NAME> (where “x” is an integer number)
Put here all factors/conditions involved in the experiment. The factor of interest must be the last and only can have a contrast of two levels or conditions. The first sample that is going to be processed must have the control/baseline/untreated level. All these factors are processed by “DESeq2” package to perform differential expression analysis using the following R design formula: F1 + F2 + ... + Fn, being Fn the factor of interest of the experiment. “F0” factor number is used to disable factors from being processed by the pipeline, i.e. any factor beginning with “F0-<NAME>” won’t be included in DESeq2 design formula.
At least two samples per level within the Factor of interest are required to perform a Differential Expression analysis.
Contains all shell script variables required to configure the pipeline. By default user-settings are configured for a paired-end reads using 20 threads.
THREADS=20 #Set max threads used.
SORTMERNA_MEMORY=5 #Set max GB of memory used by SortMeRNA (use “MAX” allows 11.7GB)
Run
To perform a functional analysis one just need to run:
./metatrans
Note: if you wish to re-run the QC analysis on a previously-parsed sample, you must first remove the QC folders (m1-log,m1-temp,m1-output) only for that sample before running the pipeline again.
For a detailed description of all MetaTrans options run:
./metatrans -h
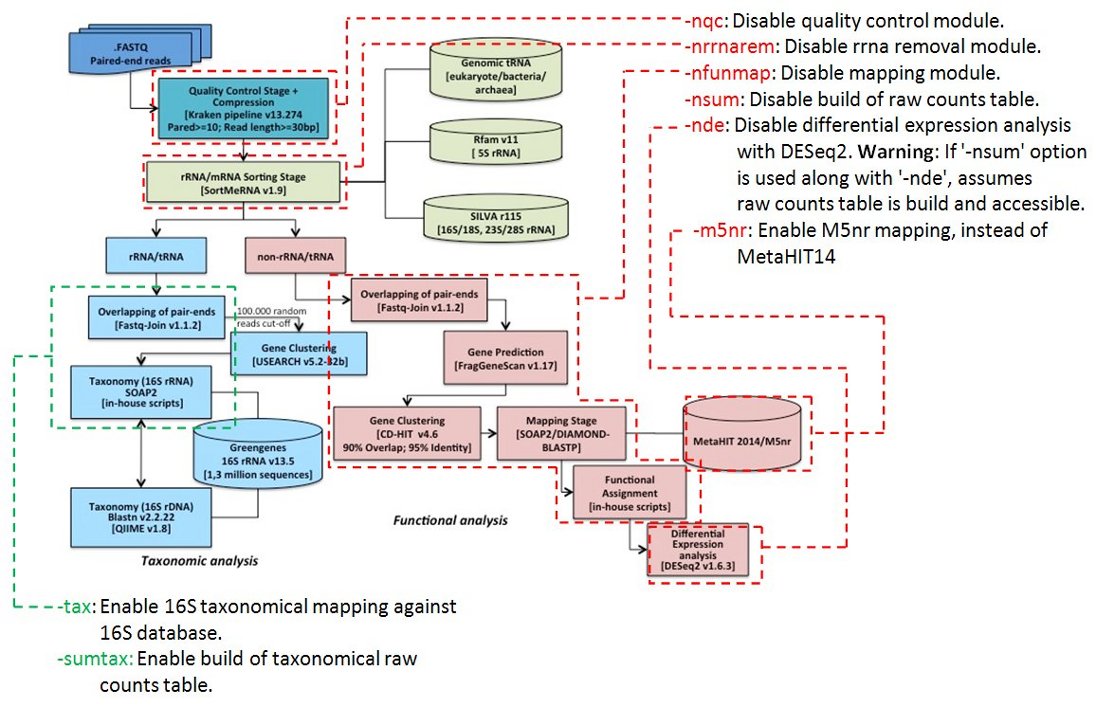 Flow diagram of the metatranscriptomic pipeline
Flow diagram of the metatranscriptomic pipeline
Output
Raw counts and differential expression files produced by the taxonomical, functional and differential expression analysis can be found here “./5-OUTPUT”:
MetaTrans-log/: output and error messages produced by the pipeline at each run. Check “Troubleshooting” section to know more on how to find more detailed information on errors.
FunctionalAbundance/MetaHIT14-EggNOGv3/: functional abundance summary produced by all samples of the experiment in MetaHIT14-EggNOGv3.
FunctionalAbundance/M5nr/: functional abundance summary produced by all samples of the experiment in M5nr-EggNOG.
FunctionalAbundance/DESeq2-Analysis/funcat/: differential expression analysis produced at a functional category level (normalised counts, statistical test results, up/down regulated functions, PCA plot and others).
FunctionalAbundance/DESeq2-Analysis/orthids/: differential expression analysis produced at orthologous ids (EggNOGv3) level (normalised counts, statistical test results, up/down regulated functions, PCA plot and others).
TaxonomicalAbundance/Greengenes135/: taxonomical abundance summary produced by all samples of the experiment (only available when supplying “-tax -sumtax” options to the pipeline”). A summary table is produced for each of the 7 taxonomical ranks: Kingdom(k), Phylum(p), Class(c), Order(o), Family(f), Genus(g), Species(s).
In turn, each module of the pipeline also produces these three folders (X is the module number: 1,2,3):
mX-log/: keep logs of all tasks involved in the module
mX-temp/: keep temporal files that could be used during execution
mX-output/: keep results files produced by the module. In the case of the quality control module, several reports are created within these folders:
report_fastqc_raw/: FastQC initial report
report_fastqc_after_trimming/: FastQC report produced after trimming stage
report_qc_reaper_after_trimming/: reaper QC report after trimming stage produced by “Kraken tools”
report_qc_after_collapse_and_filter/: QC report created after collapse+filter stage produced by “Kraken tools”
Uninstallation
Just remove the MetaTrans root folder where you unpacked all files.
More in detail
Additional configuration
ByThe following sections describe more in detail some files and aspects of the pipeline that might be customized to better fit particular needs.
Paired-end samples without barcodes scenario
Please note that the variables described within “settings.txt” file should be configured for each study in order to perform the quality control step using the Kraken tool, as it depends on the adaptors used when sequencing.
See here for a detailed description of this scenario and here (section "Paired end RNA sequencing") for a worked example of a pair-end scenario in Kraken tools.
- FIVEPADAPTOR, THREEPADAPTOR:
These two variables must be filled out with the adaptor sequences corresponding to the 5' adaptor and 3' adaptor, in sense orientation. Kraken will use these two sequences to trim/filter adaptors in pair-end1 reads, and will automatically complement and swap these sequences to find adaptors in the pair-end2 reads (see here in "5p_ad and 3p_ad" section for more detailed info).
Pair-end1 and pair-end2 reads within fastq files are assumed to be given in sense orientation (5' -> 3'). The 5' adapter will be treated as a “tabu” sequence, i.e. if present in a read the read will be discarded. There is currently no provision to simply remove the matching part. Conversely, the 3' adapter will be trimmed from the read if present.
If you do not know the 3' adapter it is possible to search for it in the input files with the program minion, which is shipped with reaper. For a FASTQ file called input.fq.gz usage is as follows:
./0-Software/thirdpartytools/Kraken-13-274/kraken_tools_binaries/minion search-adapter -i input.fastq
By default minion will take the first 2 million reads. Use the “-do” option to change this.
E.g.:
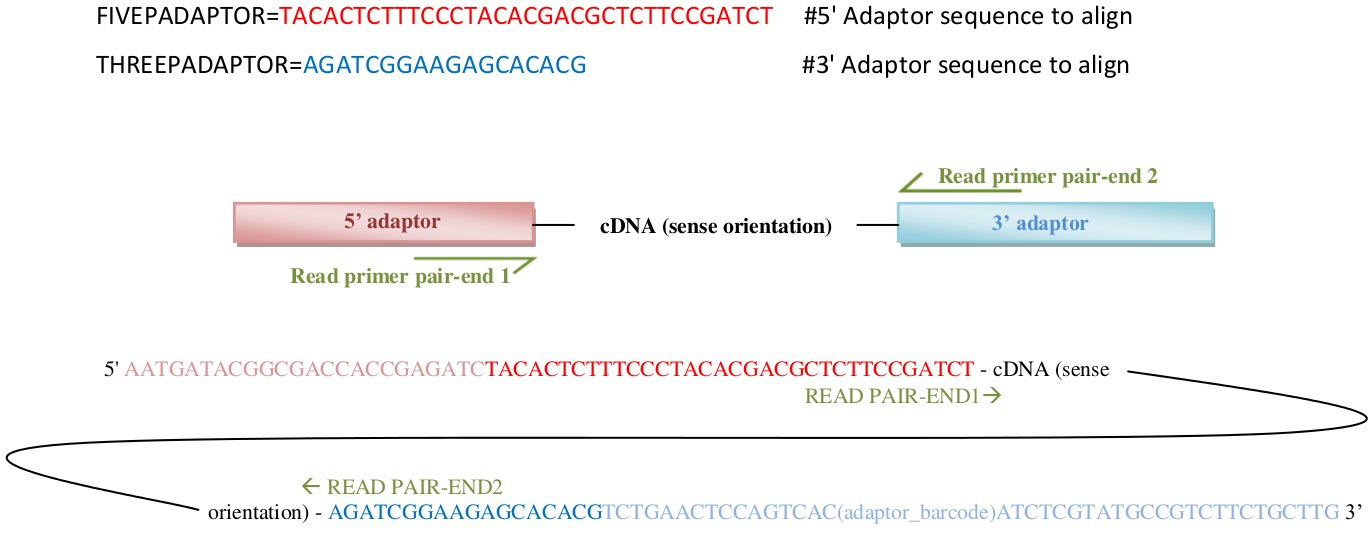 Brief description of adaptor's sequences
Brief description of adaptor's sequences
- TRIMSETTINGS:
A full description of all these options for the trimming/filtering process by means of the "reaper" tool is provided by Kraken tools here. The following descriptions are partially adapted from the original documentation.
-geom no-bc (reads geometry)
Describe the geometry of reads in fastq files. This geometry is already configured for a paired-end scenario.
-3p-global 14/2/0 (3’ alignment)
Specifies best alignment found anywhere between the read and 3' adapter. Any stretch of 14 bases with at least 12 bases matches, a maximum of 2 mismatches and no gaps (indels), between 3' adapter and the read, will be considered a match. Find further information about the alignment format below.
-3p-prefix 8/2/0/0 (3’ alignment)
This option specifies stringency criteria for an alignment that matches the start of the 3' adapter with the end of the read. Find further information about the alignment format below.
-3p-head-to-tail 0 (3’ alignment)
This option specifies the minimum length at which a perfect match at the end of the read to the beginning of the 3’ adapter should be trimmed. It is worthwhile if reads are heavily contaminated with 3' adapter sequence.
-mr-tabu 14/2/1 (5’ alignment)
Match requirements for tabu sequences (sequences containing 5' adaptor). If a match is found anywhere having at least 12 bases matches, a maximum of 2 edits (mismatch/gap) and no more than one gap (indel) between 5' adapter and the read, the read is discarded. There is currently no provision to simply remove the matching part. Find further information about the alignment format below.
-clean-length 30 (length-based filtering)
After adapter matching and trimming the length of a read may be short. With -clean-length 30 any read shorter than 30 will be discarded.
-nnn-check 3/5 (N-masked bases)
Bases may not be called and show up as N in the read. This trimming test uses a sliding window to identify whether a read suffix should be removed. Any sliding window of 5 bases containing at least 3 Ns will be trimmed along with the remaining suffix of the read.
-qqq-check 43/5 (low quality check)
Low quality sequence can be detected using the median quality value in a sliding window. The quality-based trimming test is defined by the first base where this median value drops below a specified threshold. This test is applied to the full read unless an extra "/<offset>" argument is specified (see here for further documentation):
-qqq-check <cut-off>/ <length>/<offset>
The cutoff relates to the raw ASCII values found in the file and expressly not to the transformed P-values they represent (phred quality scores).
E.g.: to set a quality Phred score cut-off of 10 in Illumina 1.8+, the value would be: 33+10 = 43
See here for further info on fastq phred scores.
Alignment format
The options -3p-global, -3p-prefix, -3p-head-to-tail, -mr-tabu use an alignment format that describe the conditions that will tell if the alignment is a match or not:
l/e[/g[/o]]
mismatch=substitution=base changed by another
gap=indels (insertion or deletion)
edit=mismatch or gap
l <int> minimum length required to count sub-alignment as match. The subpart stretches over at least l bases.
e <int> maximum allowed edit distance. There are no more than e edits in the subpart.
g <int> [optional, not active when set to 0] maximum allowed number of gaps. The total gap length in the subpart does not exceed g.
o <int> [optional, not active when set to 0] offset:
o= 5 requires alignment to start in the first five bases of adaptor.
o=-5 requires alignment to end in the last five bases of adaptor.
The match occurs within an offset of o bases. The exact meaning depends on the part being matched. A zero value implies on offset requirement at all.
- FILTERSETTINGS:
All these settings are disabled by default with "NA" (not available) value, but can be set to perform further filter or trimming. A detailed description of these options can be found here in the "filter" section.
FILTERSETTINGS="low NA
minSize NA
maxSize NA
five NA
three NA"
If your reads are not paired-end, the MetaTrans pipeline shell scripts should be adapted to the new scenario (the geometry of the reads would be different and should be changed for the Kraken pipeline according to this documentation).
FastQC performs detailed QC reports from raw raw sequence data coming from high throughput sequencing pipelines. For the identification of contaminant sequences it uses the following file that can be customized:
“./0-Software/config/fastqc/contaminant_list_customized.txt” which describes all contaminant sequences that will be identified in the “FastQC” reports. We can customize this file and add our specific adaptor sequences (<TAB> tabular character):
FivePrime Adaptor (custom) <TAB> TACACTCTTTCCCTACACGACGCTCTTCCGATCT
TrheePrime Adaptor (custom) <TAB> AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC
…
Kraken tools quality control pipeline
For the quality control module the pipeline use an specific quality control pipeline named “SequenceImp” belonging to the “Kraken tools” suit. This pipeline, when used for quality control, is mainly conformed of two stages: trimming stage (namely “reaper”), which uses a program named “reaper”, and a collapsing and filter stage (namely “filter”), which uses a program named “tally” to collapse reads and perform some of the filters. Three files are required to control the pipeline: a metadata file that describes the library geometry (*) of the paired-end input files, and two more files that control “reaper” (trimming/filter) and “filter” (collapsing/filter) stages.
For a further description of theses Kraken stages please check the original documentation:
-SequenceImp pipeline: here
-Reaper process: here
-Filter process: here
(*) a geometry is defined by Kraken as a description of what a read looks like, i.e. the read design.
Folder structure
The pipeline content is distributed into these folders: Databases/, Software/, Sequences/, PROCESSED_SAMPLES/ and OUTPUT/ (Fig1). Once MetaTrans finishes all sample analyses, all intermediate data that was generated by each module is saved by default into a folder for each sample. Those folders lay within PROCESSED_SAMPLES/ folder (Fig2). If “-m pipeline” option is used, a folder for each module is created instead of PROCESSED_SAMPLES/. In this case, within each module’s folder a folder for each sample is created which will only contain all intermediate data created for that module (Fig3).
![Fig1. Pipeline's folder.
Fig2. Output structure A. “-m sample [default mode]” All intermediate data generated in each module hangs within each sample name.
Fig3. Output structure B. “-m pipeline” option All intermediate data generated in each module hangs within each module name.](attachments/Image/image7.jpeg) Fig1. Pipeline's folder.
Fig2. Output structure A. “-m sample [default mode]” All intermediate data generated in each module hangs within each sample name.
Fig3. Output structure B. “-m pipeline” option All intermediate data generated in each module hangs within each module name.
Fig1. Pipeline's folder.
Fig2. Output structure A. “-m sample [default mode]” All intermediate data generated in each module hangs within each sample name.
Fig3. Output structure B. “-m pipeline” option All intermediate data generated in each module hangs within each module name.
Troubleshooting
Finding the source of the error:
In case of pipeline’s errors, to trace back to the error source, one should check first the content of the last MetaTrans log in “5-OUTPUT/MetaTrans-log” (sort by “Date Modified”). To find out the meaning of some common shell error numbers (e.g. “#ERROR# Pipeline exited with error code 127”) one might check this table or this other information on exit status.
If the error number is not helpful, then the next step is to identify last log files produced by the last module that was running when the error occurred, e.g.: "1-PROCESSED_SAMPLES/C1JB3ACXX_1_3/3-MAP/m3-log" (log files produced by module 3 in the sample with name “C1JB3ACXX_1_3”). Sort files by last “Date Modified” and check last logs.
If the error comes from the differential expression analysis, logs are placed in: “5-OUTPUT/MetaTrans-log/*.R.log”
Re-running the pipeline after the error:
To re-run the pipeline when an error occurs, first identify from the output which sample failed, then go to "1-PROCESSED_SAMPLES/<SAMPLE_NAME>/" folder and delete the entire folder of that module (e.g: “1-QC”,”2-rRNA-removal”…). In case only failed “DE” analysis, the folder to be removed is: “5-OUTPUT/XXXAbundance/XXX/DESeq2-Analysis/”.
Then modify the metadata file to run only the sample that failed (in the “Order” column set other samples to “0”) and rerun “./metatrans” using the options (./metatrans -h) to disable all modules but the failing one.
In case the job performed for that particular module lasted too much time as to be entirely repeated, you may tweak the metadata file and the shellscript of the respective module to run the module just from a certain task for that particular sample (just comment out the lines of those already finished functions in the bottom of the shellscript).
Further questions or comments might be sent to :
metatrans-forum<at>googlegroups.com